Home
You'll need a git client, Python, and a couple Python packages. On an Ubuntu Linux system, this is quickly accomplished by running:
sudo apt-get install git python python-matplotlib python-numpy python-scipy
On other operating systems, I find that using easy_install is the best bet for getting Python packages. Install easy_install, then get numpy, scipy, and matplotlib.
In development I use python 2.7.3, scipy 0.9.0, numpy 1.6.1, and matplotlib 1.1.1rc.
Now, grab the development snapshot by running:
git clone https://github.com/matted/multipool.git
You could also skip the git step by downloading the current version of mp_inference.py directly from the web interface.
Here are some quick usage examples based on the example files included in the software distribution:
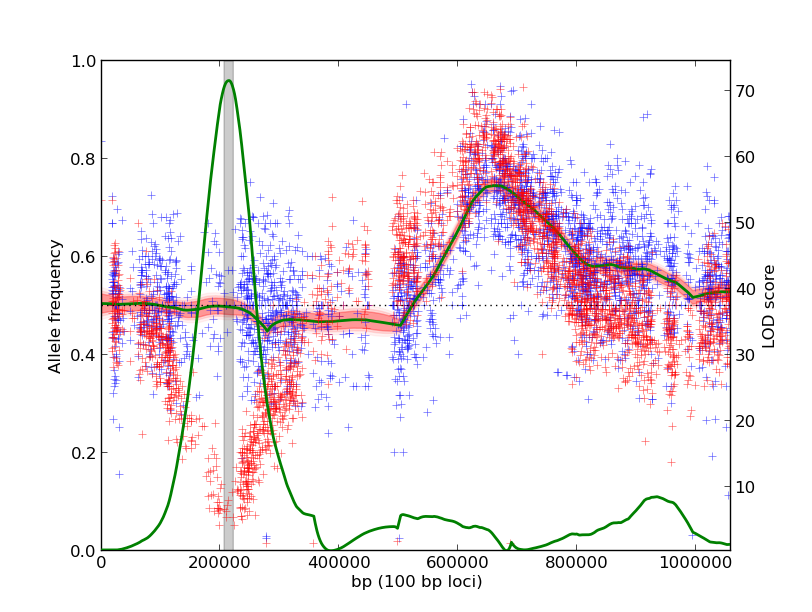
-Compare two experiments for significant differences. Here, the null hypothesis is that the underlying allele frequencies across the genome are the same. Departures from this assumption are scored for significance. This is useful for comparing a selection against a null experiment or opposite phenotypic extremes.
./mp_inference.py -n 1000 poolK1_chr12.txt poolK2_chr12.txt -m contrast
The output plot identifies a locus on the left arm of the chromosome that is different in the two experiments:
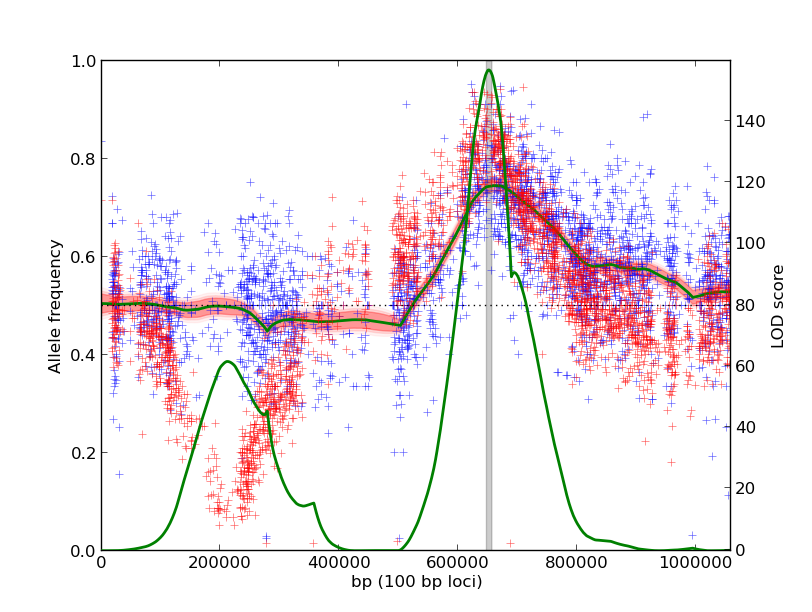
-Leverage multiple experiments as biological replicates. Here, the null hypothesis is that the underlying allele frequencies across the genome are 50%, suggesting no correlation with the phenotype. The alternate hypothesis is that the replicate experiments have the same, non-50%, allele frequency. Likelihood ratios comparing these hypotheses are computed across the genome.
./mp_inference.py -n 1000 poolK1_chr12.txt poolK2_chr12.txt -m replicates
Using the same input data as the first example, we identify a shared QTL on the right arm of the chromosome. The QTL on the left arm is not as significant because the two experiments have dramatically different underlying allele frequencies.
When interpreting the results, a careful consideration of the modeling assumptions is necessary. From current experience, the following assumptions are the most important:
- Uniform recombination rate across the genome, and in particular, equal around a QTL peak
- Uncorrelated errors in allele frequency noise
- Equal DNA representation from each member of the pool in the sequenced library
- Relatively accurate allele frequency calls for the input count data
Current work is aimed at reducing the dependence on these assumptions. One workaround is to reduce the pool size when computing peak intervals, since this approximately reflects the additional noise from e.g. pooling heterogeneity.
- How can I learn more about the details and motivations behind Multipool?
For now, the best bet is to read the paper. As this wiki grows, it will become a better resource for this task.
- How do I report a bug in the software or request a clarification in usage?
The quickest way currently is to email Matt Edwards. You can also add an issue through Github, which I'll use to track feature requests and bug reports.