NAMD Protein Ligand Complex Simulations
Choose the protein of choice with ligand in it. In this example 4W52.pdb is used. It is a structure of T4 Lysozyme L99A with Benzene Bound. Since this structure has only one chain, nothing has been done. If there are more than one chain, keep only chain A/B to do MD simulations or FEP simulations further.
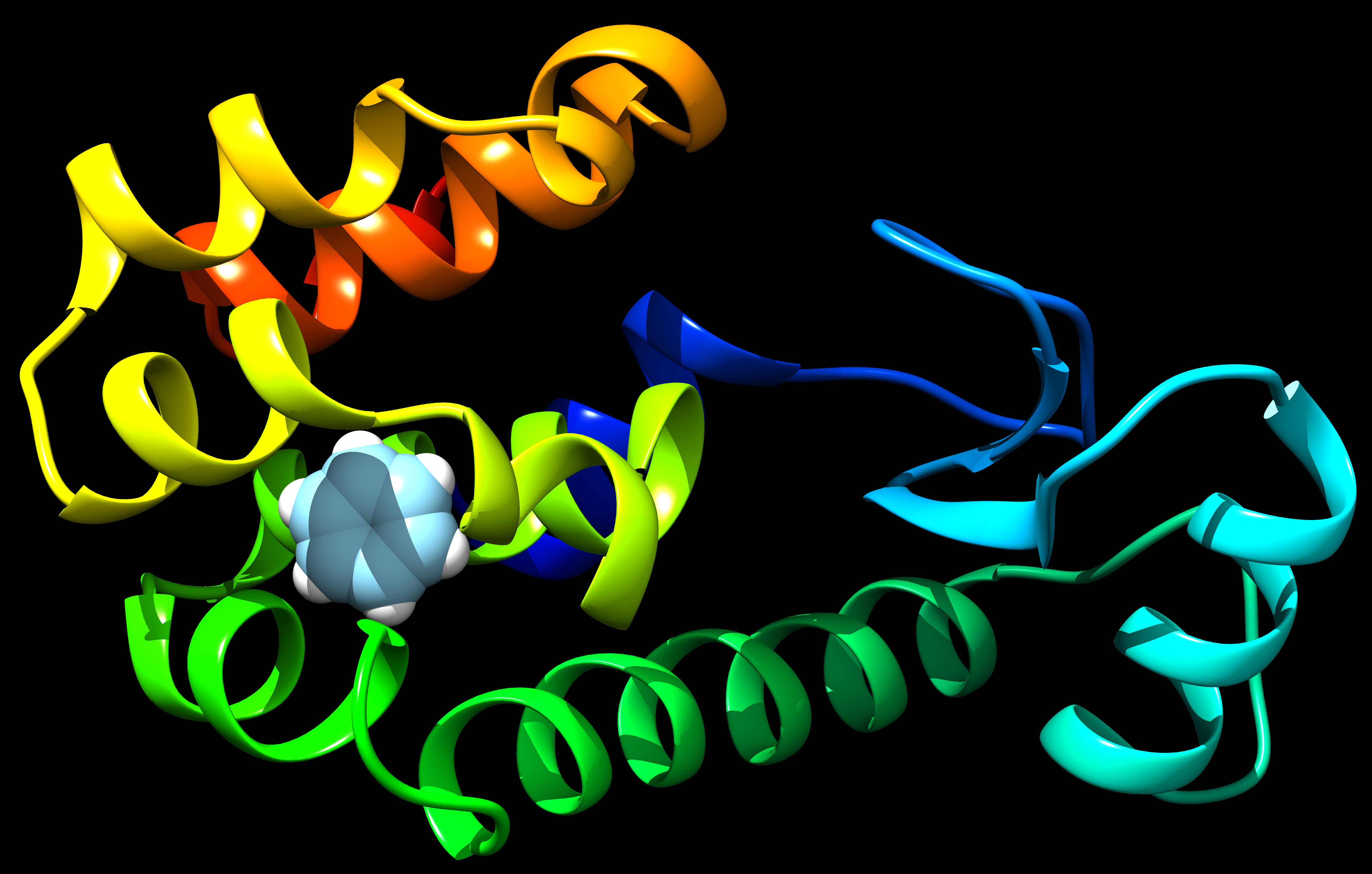
Code below uses dockprep functionality of Chimera to clean the protein and to add missing residues using Dunbrack Library. Hydrogens are not added to the protein as it was done automatically by pdb2gmx and to avoid any issues with differences in atom names. Hydrogens are added to Ligand as we have forcefield parameters explicitly defined for ligand atom names.
## PDB_FILE SHOULD THE COMPLETE PATH OF THE FILE
## REPLACE BNZ with LIGAND resname
## USAGE: Chimera --nogui --script "prep_prot_lig.py 4w52.pdb BNZ"
import chimera
from DockPrep import prep
import Midas
import sys
import os
PDB_file = sys.argv[1]
lig_name = sys.argv[2]
os.system('grep ATOM %s > %s_clean.pdb'%(PDB_file,PDB_file[:-4]))
os.system('grep %s %s > %s.pdb'%(lig_name,PDB_file,lig_name))
protein=chimera.openModels.open('%s_clean.pdb'%PDB_file[:-4])
ligand=chimera.openModels.open('%s.pdb'%lig_name)
prep(protein,addHFunc=None,addCharges=False)
prep(ligand)
Midas.write(protein,None,"protein_clean.pdb")
Midas.write(ligand,None,"ligand_wH.pdb")Run this code by using Chimera. Make sure you have Chimera installed and can be called from Command Prompt.
Chimera --nogui --script "prep_prot_lig.py 4w52.pdb BNZ"
This produces protein_clean.pdb and ligand_wH.pdb that will be used further. Make sure that right number of hydrogens are added and the charge on ligand matches the expected value.
Upload the ligand_wH.pdb to LigParGen server and for protein-ligand simulation, before uploading make sure that ligand residue name is changed to 1. This is because BOSS, the core of LigParGen server only works for only a certain number of residues. Download the files for CHARMM/NAMD, i.e BNZ.prm (parameter file)and BNZ.rtp (connectivity) files.
combine the PDB coordinates of apo-protein and ligand by cat protein_clean.pdb BNZ.pdb > complex.pdb and remove the connectivity information and TER. Change the residue number of Benzene ligand for continuity.
- Load
complex.pdb - Open
Extension -> Modeling -> Automatic PSF generator - Remove default Topology file.
- Download topology file for OPLS-AA/M from Jorgensen group and add the path to topology files.
- Add the path of
BNZ.rtffile too. - Click
Load input files - Click
Guess and Split Chainsit will ask you for location of complex.pdb again, so add the location - Click
Create Chains - Solvate the protein using
Extension -> Modeling -> Add Solvation boxand use PSF and PDB files generated from AutoPSF generator. - Neutralize the system using
Extension -> Modeling -> Add Ions
If you choose to do it using command line VMD. Use the code below
package require psfgen
topology top_opls_aam.inp
topology BNZ.rtf
pdbalias HIS HSD
pdbalias atom SER HG HG1
pdbalias residue HIS HSE
pdbalias atom ILE CD1 CD
segment A {pdb complex.pdb}
coordpdb complex.pdb A
guesscoord
writepdb complex_autopsf.pdb
writepsf complex_autopsf.psf
package require solvate
solvate complex_autopsf.psf complex_autopsf.pdb -t 5 -o complex_wb
package require autoionize
autoionize -psf complex_wb.psf -pdb complex_wb.pdb -neutralize -o ionized
set ubq [atomselect top all]
measure minmax $ubq ## USE THE DIFFERENCE BETWEEN MIN AND MAX FOR CELL BASIS VECTOR DIMENSION
measure center $ubq ## USE THE CENTER FOR CELLBASIS ORIGIN
exit
Run it using $VMD -dispdev text -e UNK.pgn and it will generate ionized.psf and ionized.pdb files that will be used for performing MD simulations.