This tool was developed as part of Gardner et. al., Contribution of Retrotransposition to Developmental Disorders, 2019. It uses discordant pairs and split reads to identify processed psueudogenes in individual human genomes from whole exome sequencing data (WES). The tool is coded entirely in Java v1.8. The tool runs in three steps with each, along with examples, included in sections following this introduction and basic use instructions.
This repository only contains source code. Please see Releases to download a functional .jar.
This section details basic information required by the Retrogene discovery pipeline.
| Option | Required? | Description |
|---|---|---|
| b | YES | path to the bam file being analyzed. |
| h | YES | path to the reference genome. Reference must be indexed with the same path as the reference but with .fai suffix. |
| o | YES | output file for this particular step. Note: please make sure to change this path between steps or you could risk overwriting previous files! |
| a | YES | path to output file from 'Aggregate' step (see below for more details). |
| l | YES | file of files provided as input to 'Aggregate' or 'Merge' step (see below for more details). |
| g | YES | Gene file with one line per transcript (see below for more details). |
| k | NO | List of known pseudogenes from other literature (see below for more details). |
| p | NO | pLI and gene name annotation for all genes in -g (see below for more details). |
Example gene files for build GRCh37 of the human reference genome are provided in the /ReferenceFiles/ folder on this repository. Only the gene annotation file provided to -g is required by Retrogene. Explanations for each are below:
-g:
Follows the .bed12 format, with one line for each transcript, except for the last column, which is a unique identifier that links transcript lines together under one ID. The current files use ENSEMBL identifiers but can be switched to anything that provides a unique identifier in column 4, and an identifier that links transcripts together in column 13.
-k:
No required by default. This is just a list of genes, one per line, that have been previously known to have cause a processed pseudogene. These IDs have to be identical to those provided in column 13 of the file provided with -g. Simply used for annotation during Discover/Genotype and does not effect discovery/genptyping.
-p:
Not required by default. Probability of being loss of function intolerant (pLI; Samocha et. al. 2015, A framework for the interpretation of de novo mutation in human disease, Nature Genetics, 2014) annotations for all genes listed in column 13 of the -g file. Columns listed are:
- Primary Transcript
- GeneID (must be identical to
-gcolumn 13) - Gene name
- Chromosome
- Gene start
- Gene end
- pLI score
While not required, it is highly recommended to include, as this file provides a decent amount of annotation to the overall pipeline (gene name, primary transcript, pLI score). However, it is only for annotation during Discover/Genotype and does not effect discovery/genotyping.
Note: By default, the pipeline avoids performing any discovery on any gene which matches /HLA/.
Retrogene uses the Java virtual machine (v1.8) to run. The basic use is:
java -jar Retrogene.jar <Runtime>
Where runtime is one of:
| Runtime | Description |
|---|---|
| Discover | Discover processed pseudogenes in a single bam file. |
| Aggregate | Aggregate discovered genes across samples to determine presence/absence during Genotype. |
| Genotype | "Genotype" (actually determine presence/absence) of each gene identified during Discover in a single individual. |
| Merge | Merge genotyped samples together into a final file format. |
A complete run of Retrogene involves each of the three steps listed in the table above. Complete instructions are as follows:
Discover processed pseudogenes in a single bam file. Example command line:
java -jar Retrogene.jar Discover -b <bam file.bam> -h <path to reference.fa> -o <name.out> -g hg19.ensembl.bed -k KnownPS.txt -p pLI.txt
Output provided includes a lit of putative processed pseudogenes and a bed file of duplicated exons. See below for output.
Aggregate discovered genes across multiple samples:
ls *.out > file_of_files.txt
java -jar Retrogene.jar Aggregate -l file_of_files.txt -o aggregate_genes.txt -g hg19.ensembl.bed -k KnownPS.txt -p pLI.txt
Genotype an individual bam file for all genes discovered in a cohort:
java -jar Retrogene.jar Genotype -b <bam file.bam> -h <path to reference.fa> -o <name.genotyped.out> -a aggregate_genes.txt -g hg19.ensembl.bed -k KnownPS.txt -p pLI.txt
Output provided includes a lit of putative processed pseudogenes and a bed file of duplicated exons. See below for output. The 'rescue' step here involves setting a slightly lower threshold for ascertainment and additionally using SRs within ±5bps of an exon junction.
Merge genotyped samples together into a final file format (see below):
ls *genotyped.out > file_of_genotyped_files.txt
java -jar Retrogene.jar Aggregate -l file_of_genotyped_files.txt -o final_calls.txt -g hg19.ensembl.bed -k KnownPS.txt -p pLI.txt
Output for Discover takes the following format:
| Column | Description |
|---|---|
| 1 | Gene chromosome |
| 2 | Gene start coord. |
| 3 | Gene end coord. |
| 4 | Sample Name (taken from the .bam file provided with -b) |
| 5 | Gene Name (if -p file provided) |
| 6 | Gene ID |
| 7 | Number of exons included in duplication based on DRPs |
| 8 | Possible number of exons included in duplication |
| 9 | pLI score (if -p file provided) |
| 10 | Is a known pseudogene? (if -k file provided). Always false if no -k file. |
Genotype has the additional two columns attached to the end of this file as well as a column for SR totals calculated as part of genotyping:
| Column | Description |
|---|---|
| 1 | Gene chromosome |
| 2 | Gene start coord. |
| 3 | Gene end coord. |
| 4 | Sample Name (taken from the .bam file provided with -b) |
| 5 | Gene Name (if -p file provided) |
| 6 | Gene ID |
| 7 | Number of SRs across the pseudogene |
| 8 | Number of DPs across the pseudogene |
| 9 | Number of exons included in duplication based on DRPs |
| 10 | Possible number of exons included in duplication (this is a total across all transcripts, not just from one transcript model) |
| 11 | pLI score (if -p file provided) |
| 12 | Is a known pseudogene? (if -k file provided). Always false if no -k file. |
| 13 | Percent of reads that are discordant pairs in this gene (see below). |
| 14 | p-value based on the z-score of the distribution of all genes in this sample (see below). |
Merge creates a tab-delimited format with samples stored similar to VCF (i.e. columns store presence absence per individual). The file contains a header where columns are:
| Column | Description |
|---|---|
| 1 | Gene ID |
| 2 | Gene chromosome |
| 3 | Gene start coord. |
| 4 | Gene end coord. |
| 5-N | Sample-specific columns where N is the total number of samples analyzed. Presence of a duplication is indicated by a '1' whereas absence is '0' |
Column 13 and 14 of the final genotype output represent a measurement of what percent of a genes overlapping reads in the underlying WES data are discordant (i.e. map between one exon to the other). It is a decent measure of the likelihood of a gene being duplicated, but is not a perfect measure, mostly due to the length of a gene compared to the total number duplicated exons; e.g. a gene may be very long with many exons, but only the first two exons on the 3' end are duplicated, resulting in a low percent of overall reads as discordant. The overall distribution is roughly normal, and z-scores and p-values are calculated for each gene from a sample as shown in the figure below:
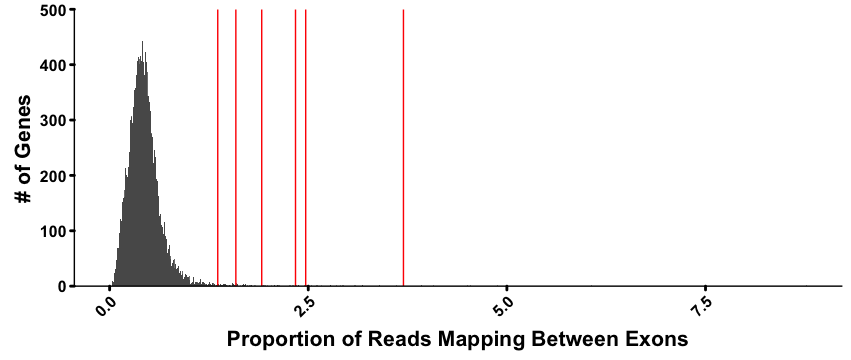 Distribution of the percent of reads which map between exons for each gene in the human genome for an individual from the DDD study. Genes which have been duplicated are marked with red lines. All have a p-value of at least 2e-4 based on the underlying distribution.
Distribution of the percent of reads which map between exons for each gene in the human genome for an individual from the DDD study. Genes which have been duplicated are marked with red lines. All have a p-value of at least 2e-4 based on the underlying distribution.
This distribution is provided as a file with suffix .dist attached to the file provided to -o during Genotype as one gene per line with gene name and % discordant pairs in column one and two, respectively. This distribution should not be used to call pseudogenes as there are a number of genes in the genome that have this quality and have not been duplicated. It should instead be used as a potential indicator of the likelihood a gene called by this pipeline is indeed duplicated.
If using this software, please cite:
Gardner et. al., Contribution of Retrotransposition to Developmental Disorders, Nature Communications, 2019
If using the list of known processed pseudogenes provided in this package, please cite the following manuscript:
Zhang et al., Landscape and variation of novel retroduplications in 26 human populations, PloS Computational Biology, 2017.