In the past few years, long-read sequencing technologies have been developed by Pacific Biosciences (PacBio) and Oxford Nanopore Technologies (ONT) (Ameur et al., 2018). PacBio and ONT technologies can generate long reads in tens of kilobase pairs, thus making it possible to obtain a complete assembly. Compared with the PacBio technology, the ONT MinION is affordable and portable and enables real- time analysis, which render it more attractive for in-field and clinical deployment.
The applications of the ONT MinION range from microbial genome assembly to cancer variant discovery and transcript isoform identification. Other studies combined both illumina generated short reads and MinION generated long-reads to assembly circular bacterial genomes. However, in the absence of short reads, MinION long-reads alone can be used to generate complete circular genomes in a rapid and cost-effective manner. Generating a complete circular bacterial genome is essential especially in monitoring and surveillance of antimicrobial resistance transmission.
Nanopore data analyses requires many bioinformatics tools to be implemented in the genome assembly pipeline. It is important to deploy the required tools in a docker image for one stop analysis for the reconstruction of a circular bacterial genome using MinION data.
This is a group 1 mini-project about Reproducing a published workflow for bacterial genome
- Collaborative on the project using git hub
- Learn from each other and carry out tasks in the event we are assigned any
- By the end of the project submit a reproducible workflow e.g. in Snakeamke
- Observe proper script documentation, should be informative
- Use visualizations
- Write a report about the mini project
- Download the sequence data programmatically using an API
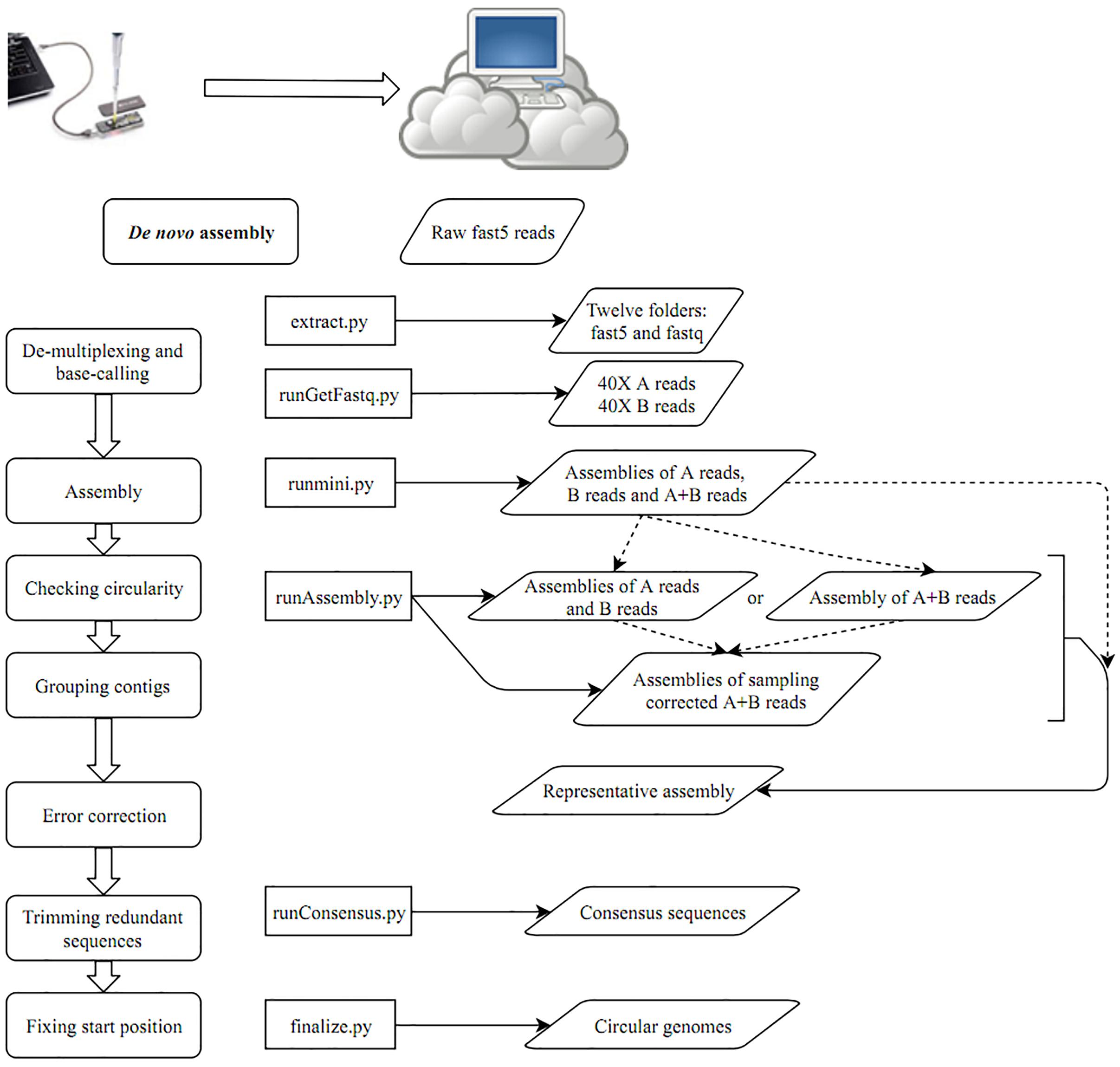
- The project uses CCBGpipe implemented in Python is available and packaged in Docker for reproducibility. Our first task is to reproduce the analysis as in the documented study README
- Reproduce the analysis workflow used for genome assembly of Oxford Nanopore MinION using Nextflow or Snakemake, and Singularity for containerization. Remember as a group we settled on Snamake workflow
Task1: Reproduce the analysis using the workflow given in the README file in CCBG_Study directory our repo
In our report, we should address the following question:
- Are we able to arrive at similar conclusions as those in the paper? Why or why not?
- What else can we glean from your re-analysis?
- Are the descriptions in the methodology section detailed for reproducibility? If not, what could we have done to improve reproducibility?
Since the data for the 12 barcodes is too large. For reproducibility, here is the link for barcode01
This was meant to reproduce the CCBGpipe workflow as done by the authors.
To make our own copy of the CCBG pipeline we clone the CCBGpipe repo as given by the authors:
git clone https://github.com/jade-nhri/CCBGpipe.git
cd CCBGpipe
Using the given docker file we built docker image for reproduction of the analysis
docker build -t "ccbgpipe:v1" ./
docker run -h ccbgpipe --name ccbgpipe -t -i -v /:/MyData ccbgpipe:v1 /bin/bash
Inside the docker: root@ccbgpipe:/#
To install java:
apt-get update
apt-get install -y software-properties-common
add-apt-repository ppa:webupd8team/java
apt-get update && apt-get install oracle-java8-installer
Please note: the Oracle JDK license has changed starting April 16, 2019.
You can download zulu to include Java (https://www.azul.com/downloads/zulu/).
apt-key adv --keyserver hkp://keyserver.ubuntu.com:80 --recv-keys 0xB1998361219BD9C9
apt-add-repository 'deb http://repos.azulsystems.com/ubuntu stable main'
echo 'deb http://repos.azulsystems.com/debian stable main' > /etc/apt/sources.list.d/zulu.list
apt-get update
apt-get install zulu-8
-
Installation of Albacore failed we therefore opted to basecall with Guppy
-
To extract fastq files using guppy_bascaller
guppy_basecaller -i path-to-raw_reads -s outpath (e.g., guppy_basecaller -i Fast5 -s guppy_out)
- To de-multiplex
guppy_barcoder -i inpath -s outpath (e.g., guppy_barcoder -i guppy_out -s barcoding)
- To produce read_id list and joinedreads.fastq for each barcode
preprocess.py -b path-to-barcoding_summary.txt -s path-to-sequencing_summary.txt -o outpath (e.g., preprocess.py -b barcoding/barcoding_summay.txt -s guppy_out/sequencing_summary.txt -o outdir)
- With the data produced by the above process, you can perform CCBGpipe by beginning with creating a Run folder
mkdir Run && cd Run
runGetFastq.py path-to-fast5 (e.g., runGetFastq.py ../outdir/)
runmini.py
runAssembly.py
runConsensus.py path-to-fast5 (e.g., runConsensus.py ../fast5/)
finalize.py outpath (e.g., finalize.py ../results)
We were able to make a snakemake pipeline up to the preprocess step.
See the snakefile for the same in the task 2 directory.
We were not able to incorporate the subsequent steps (sorting based on long-length reads, quality, assembly) in the snakefile because these steps includes multiple dependent python scripts and due to time constraints were unable to compile these scripts into modules.