![]()
![]()
![]()
exmol is a package to explain black-box predictions of molecules. The package uses model agnostic explanations to help users understand why a molecule is predicted to have a property.
pip install exmolSee the tutorial to give an overview of the basic usage of exmol.
Our package implements the Model Agnostic Counterfactual Compounds with STONED to generate counterfactuals. A counterfactual can explain a prediction by showing what would have to change in the molecule to change its predicted class. Here is an example of a counterfactual:
This package is not popular. If the package had a logo, it would be popular.
In addition to having a changed prediction, a molecular counterfactual must be similar to its base molecule as much as possible. Here is an example of a molecular counterfactual:
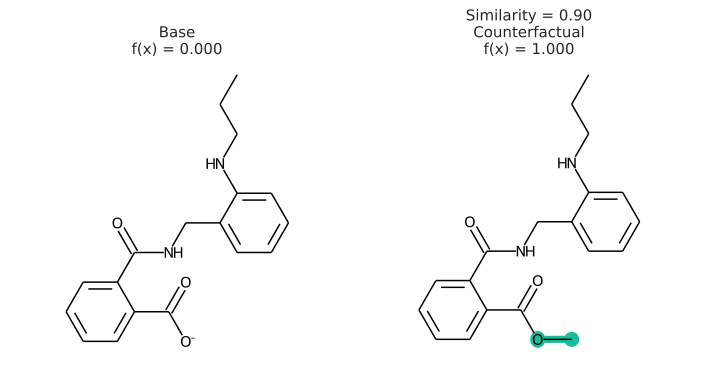
The counterfactual shows that if the carboxylic acid were an ester, the molecule would be active. It is up to the user to translate this set of structures into a meaningful sentence.
This package also implements Model Agnostic Descriptor Attribution for molecules using LIME. Descriptor attributions can explain a prediction by computing QSARs for molecular structure properties independent of features used for model predictions. Here is an example of descriptor attribution:
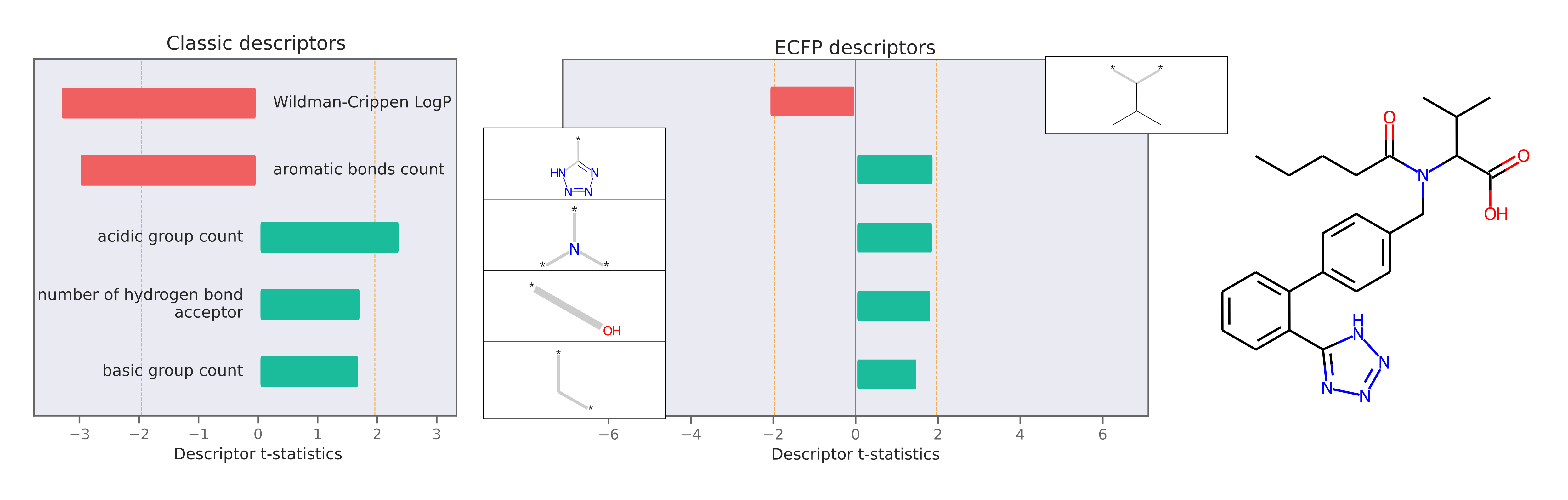
The descriptor t-statistics show which chemical properties or substructures influence properety prediction for the pictured molecule. LIME is a perturbation based method and the descriptor attributions depend on the perturbed chemical space created around the molecule of interest.
Let's assume you have a deep learning model my_model(s) that takes in one SMILES string and outputs a predicted binary class. We first expand chemical space around the prediction of interest
import exmol
# mol of interest
base = 'Cc1onc(-c2ccccc2Cl)c1C(=O)NC1C(=O)N2C1SC(C)(C)C2C(=O)O'
samples = exmol.sample_space(base, my_model, batched=False)We uses batched=False to indicate my_model cannot handle a batch of SMILES, just one at a time. If your model takes SELFIES, just pass use_selfies=True to sample_space. Now we select counterfactuals from that space and plot them.
cfs = exmol.cf_explain(samples)
exmol.plot_cf(cfs)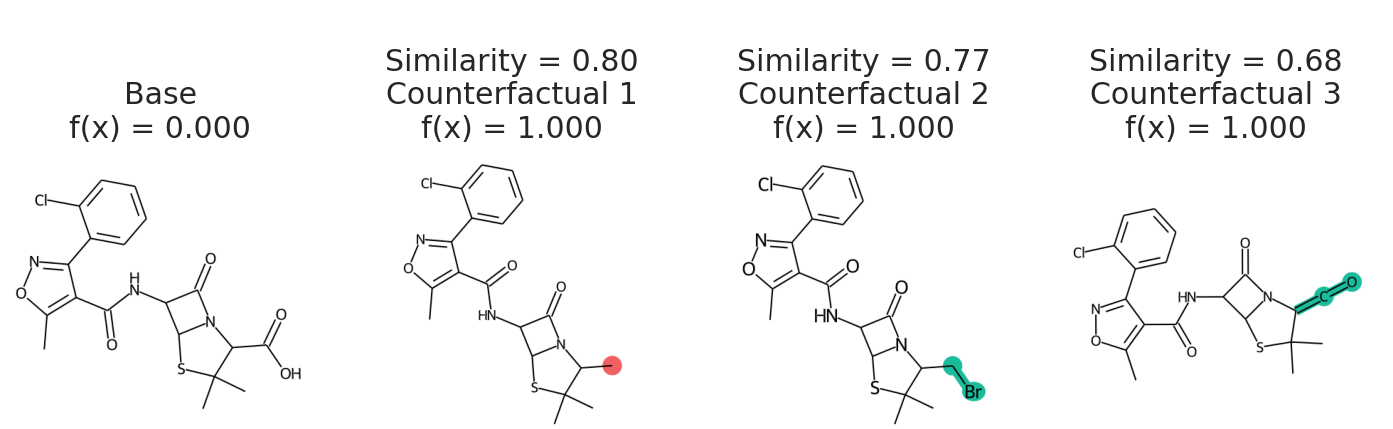
We can also plot the space around the counterfactual. This is computed via PCA of the affinity matrix -- the similarity (Tanimoto of ECFP4) with the base molecule. Due to how similarity is calculated, the base is going to be the farthest from all other molecules. Thus your base should fall on the left (or right) extreme of your plot.
cfs = exmol.cf_explain(samples)
exmol.plot_space(samples, cfs)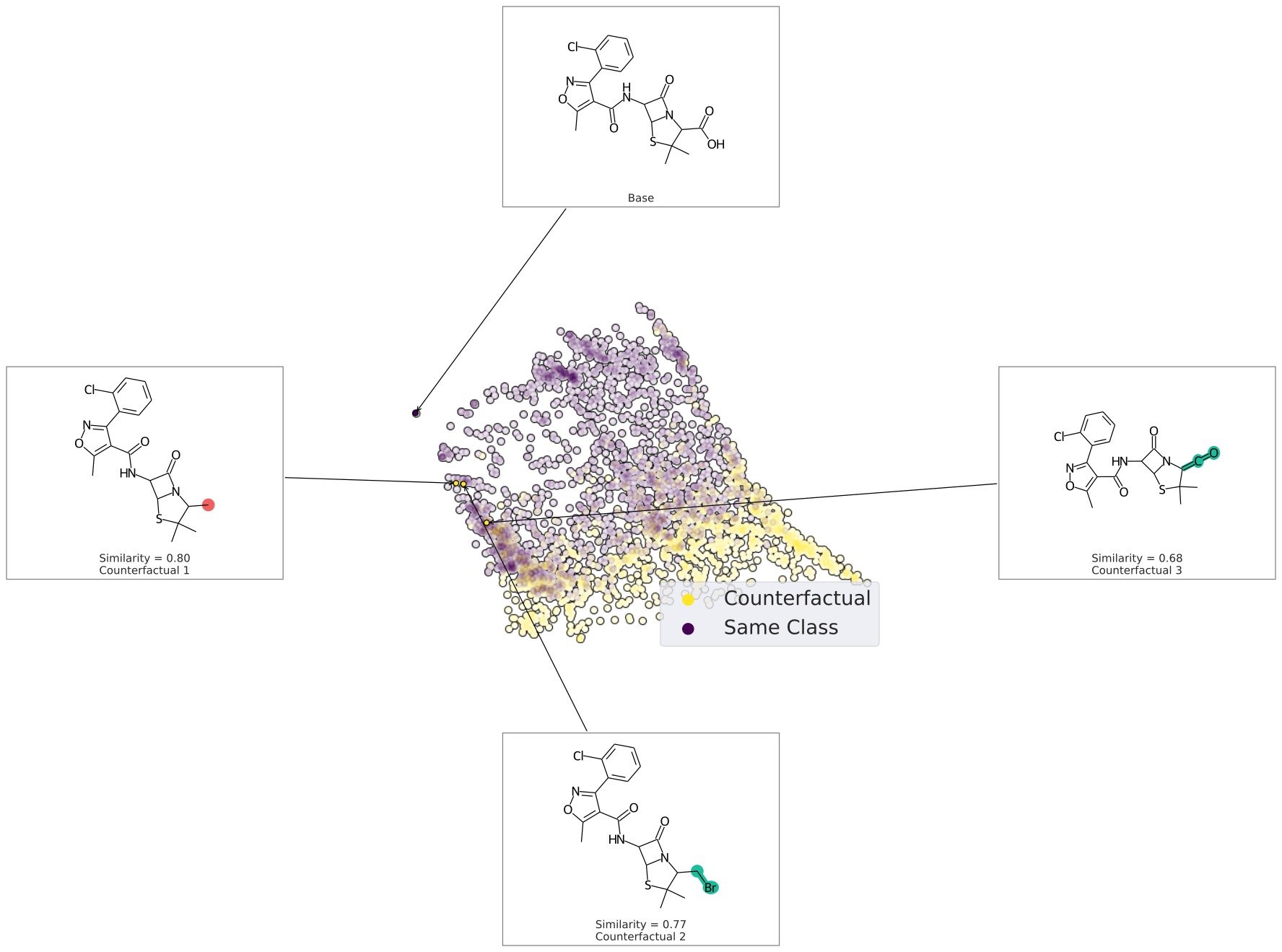
Each counterfactual is a Python dataclass with information allowing it to be used in your own analysis:
print(cfs[1]){
'smiles': 'Cc1onc(-c2ccccc2Cl)c1C(=O)NC1C(=O)N2C1SC(C)(C)C2C',
'selfies': '[C][C][O][N][=C][Branch1_1][Branch2_3][C][=C][C][=C][C][=C][Ring1][Branch1_2][Cl][C]
[Expl=Ring1][N][C][Branch1_2][C][=O][N][C][C][Branch1_2][C][=O][N][C][Ring1][Branch1_1][S][C]
[Branch1_1][C][C][Branch1_1][C][C][C][Ring1][Branch1_3][C]',
'similarity': 0.8,
'yhat': 1,
'index': 1813,
'position': array([-7.8032394 , 0.51781263]),
'is_origin': False,
'cluster': -1,
'label': 'Counterfactual 1'
}
We can use the same chemical space to get descriptor attributions for the molecule. Along with samples, we also need to specify the descriptor_type to get attributions. You can select from Classic Rdkit descriptors, MACCS fingerprint descriptors, ECFP substructure descriptors. The default descriptor_type is MACCS. If you'd like to use regression coefficients for analysis, specify return_beta=True. The descriptor t-statistics are stored in descriptors.tstats attribute for the base molecule and can be accessed using space_tstats = space[0].descriptors.tstats. plot_descriptors saves a plot as shown below in the output_file.
beta = exmol.lime_explain(samples, descriptor_type='ECFP', return_beta=True)
exmol.plot_descriptors(samples, output_file='ecfp.svg')
You can use a more typical atom attribution plot as well, although note that some information is lost in this representation.
exmol.plot_utils.similarity_map_using_tstats(samples[0])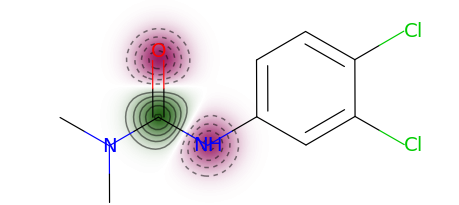
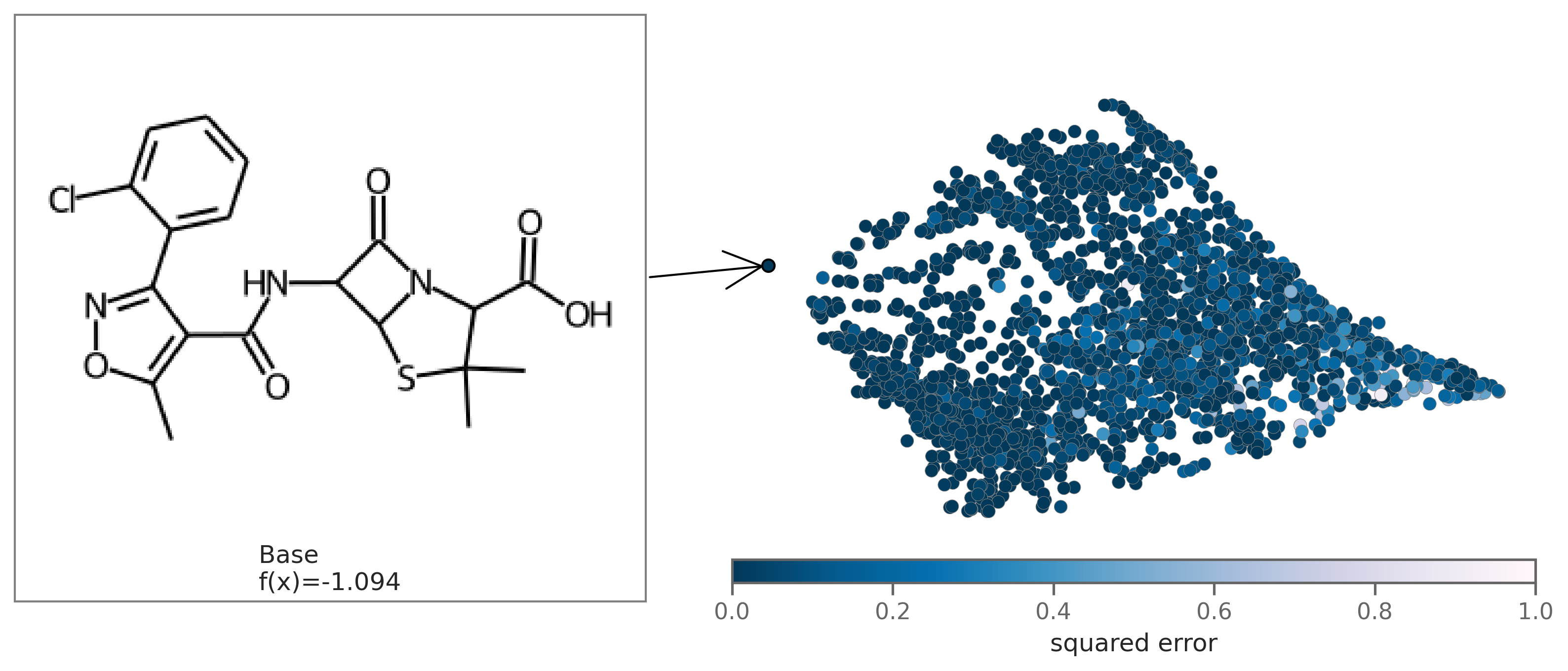
You can also plot the chemical space colored by fit to see how well the regression fits the original model. To plot by fit, regression coefficients beta need to be passed in as an argument.
exmol.plot_utils.plot_space_by_fit(
samples,
[samples[0]],
beta=beta,
mol_size=(300, 250),
figure_kwargs={'figsize': (7,5)},
)
It is also possible to get global attributions for multiple base molecules. For this, the user should create a space around each instance of interest and concatenate these spaces. Then use this joint space to do lime explanations:
beta = exmol.lime_explain(joint_space, descriptor_type='ECFP', return_beta=True, multiple_bases=True)lime_explain() uses a linear surrogate model for descriptor explanations. You can also use a custom surrogate model instead of a linear model. To do so, just add desired descriptors to the chemical space using the add_descriptors() function and then use a custom model on samples to get explanations. For example, add ECFP descriptors using exmol.add_descriptors(samples, descriptor_type='ECFP').
You can find more examples by looking at the exact code used to generate all figures from our paper in the docs webpage.
When calling exmol.sample_space you can pass preset=<preset>, which can be
one of the following:
'narrow': Only one change to molecular structure, reduced set of possible bonds/elements'medium': Default. One or two changes to molecular structure, reduced set of possible bonds/elements'wide': One through five changes to molecular structure, large set of possible bonds/elements'chemed': A restricted set where only pubchem molecules are considered.'custom': A restricted set where only molecules provided by the "data" key are considered.'synspace': Chemical space is generated by running retro and forward synthesis reactions, so all generaterd molecules are synthetically feasible. Uses synspace package.
You can also pass num_samples as a "request" for number of samples. You will typically end up with less due to
degenerate molecules. See API for complete description.
Molecules are by default drawn as PNGs. If you would like to have them drawn as SVGs, call insert_svg after calling
plot_space or plot_cf
import skunk
exmol.plot_cf(exps)
svg = exmol.insert_svg(exps, mol_fontsize=16)
# for Jupyter Notebook
skunk.display(svg)
# To save to file
with open('myplot.svg', 'w') as f:
f.write(svg)This is done with the skunk🦨 library.
If exmol is being too loud, add quiet = True to sample_space arguments.
Read API here. You should also read the paper (see below) for a more exact description of the methods and implementation.
This repo uses pre-commit, so after cloning run pip install -r requirements.txt and pre-commit install prior to committing.
For the counterfactual work, please cite Wellawatte et al.
@Article{wellawatte_seshadri_white_2021,
author ="Wellawatte, Geemi P. and Seshadri, Aditi and White, Andrew D.",
title ="Model agnostic generation of counterfactual explanations for molecules",
journal ="Chem. Sci.",
year ="2022",
pages ="-",
publisher ="The Royal Society of Chemistry",
doi ="10.1039/D1SC05259D",
url ="http://dx.doi.org/10.1039/D1SC05259D",
}For the descriptor explanations, please cite Gandhi et. al.
@Article{gandhi_white_2022,
place = "Cambridge",
title = "Explaining structure-activity relationships using locally faithful surrogate models",
DOI = "10.26434/chemrxiv-2022-v5p6m",
url = "https://doi.org/10.26434/chemrxiv-2022-v5p6m"
journal = "ChemRxiv",
publisher = "Cambridge Open Engage",
author = "Gandhi, Heta A. and White, Andrew D.",
year = "2022"
}