No DMFT convergence for the 2D Hubbard model on a TB lattice at low T? #6
Comments
|
Hi! My guess is that for the parameter regime you are considering the solution of the model is an insulator. The problem as you decrease the temperature, is that the system really wants to spin polarize. You can check this by comparing real and imaginary parts of both the 'up' and 'down' spin Green's functions. One way out it to enforce the up-down symmetry at every DMFT loop. Hope this helps! |
|
Sorry, I just realized you already impose the up/down symmetry in the script. You can try to add the following global move option in the global_moves = [(0.05, lambda (a,alpha,dag) : ( {'up':'down','down':'up'}[a],alpha,dag ) )]
|
|
Thanks, Michel! I'll try that and get back to you! |
|
Hi Michel, this improved the convergence quite a bit, as you can see from the figure (this run is with half the number of cycles used for the above): However, I don't understand why this works. Is there any documentation for this option to help me understand what it does? (I googled it and couldn't find any) |

|
There is no documentation on the global moves for now. The reason is that I think global moves can lead to misleading results and as such I consider it an "expert" feature which I did not want to expose to everybody. I will probably add an "expert" chapter somewhere in the doc. Let me try to explain what global moves are and why they can be a bit dangerous. The problem you were having is an ergodicity problem. Basically, during the Monte Carlo process, you were sampling only a smaller part of the configuration space. Because of this, configurations were not spending equal time in "up" and "down" space and you were getting polarized solutions. The origin of the ergodicity problem is that configurations are only changed locally so that you make rather small moves in the phase space. Global moves can be a way out of this. They introduce global moves that change configurations a lot allowing you to have a better sampling of the phase space. However, when your global move corresponds to a symmetry of the Hamiltonian, you somehow change a configuration to its mirror image with inverted spins. If you had an important ergodicity problem, you might still have it with global moves. Indeed, instead of sampling an island in phase space, you will sample two mirror islands in phase space. Of course, as a result, you will get unpolarized solutions. But you cannot be sure you escaped you ergodicity problem. You have to check this explicitly by changing random seeds and seeing that the final result does not change. To make a long story short, when you do not use global moves and find polarized solutions, you know that you have an ergodicity problem and you can start thinking how to fix it. If you have global moves all the time, you might not notice you have ergodicity problems because your solutions look fine (unpolarized). So I recommend you always start with no global moves, so that you know you have to be very careful in checking your results after you introduce them. |
|
Hello, Many thanks for the explanation! I tried a number of different random seeds and the imaginary part of the self-energy seems to converge to the same value for all of them. However, I also thought of checking the real part of the self energy and it seems that even with the global updates it does not converge (this is only for the lowest Matsubara frequency; at higher Matsubara frequencies it converges fine). For example: My experience (and that of some colleagues) with other DMFT solvers indicates that there's no fundamental reason why this should happen in this parameter range. I want to try and resolve that issue, or at least try to understand why it appears. I'll probably have a look at the TRIQS code to try and understand how the moves work. Petar |

Hello,
I started using TRIQS recently for DMFT simulations. In order to get used to TRIQS, I've been doing some tests with the 2D Hubbard on a TB lattice (the code is here: https://gist.github.com/pbakalov/8729382).
At relatively high temperature (beta = 10; t =1), the DMFT loop converges to a solution without problems for values of U between 0.1 and 16.
However, when I repeat the same simulation at beta = 30, it seems that for U > ~8 the simulation does not converge. This is illustrated by the attached plot (U = 12) of the value of the imaginary part of the self-energy at the first Matsubara frequency (i omega_0 = i pi /beta) versus DMFT iteration. As the DMFT loop progresses the imaginary part self-energy at the first Matsubara frequency oscillates between approximately -50 and -200.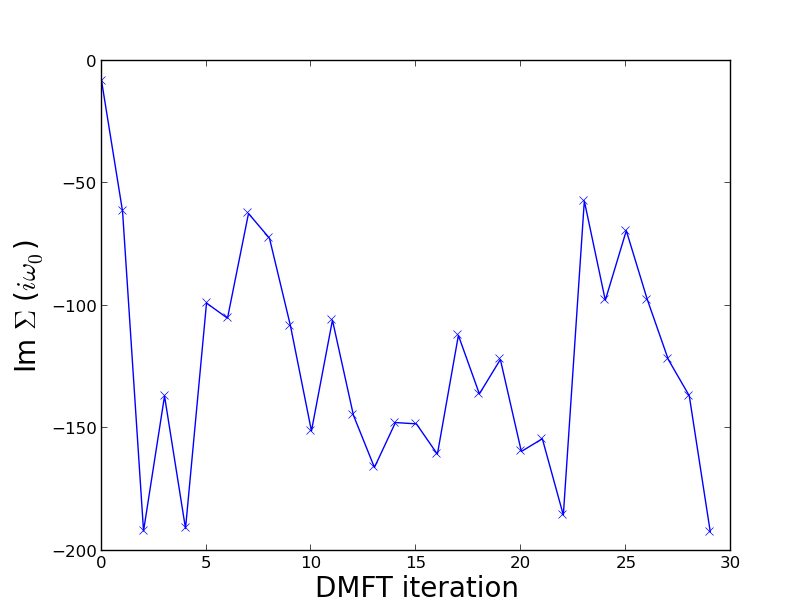
This behavior persists even when I take a fairly large number of cycles per iteration and a large number of iterations (30 cores x 1e6 cycles per core = 3e7 cycles; the cycle length is 200). For the longest run I did, I used 30 loops and 50 Legendre coefficients.
So it seems that it is practically impossible to get convergence at beta = 30 for large U. However, from colleagues who use other DMFT codes I hear that there should be no problem to reach convergence at beta = 30.
Am I missing something (say I should change the Hilbert transform parameters or the number of Matsubara frequencies I use), or is this a symptom of a bug in TRIQS?
The text was updated successfully, but these errors were encountered: